
UNOISE algorithm
See also
UNOISE paper
Should I use UPARSE (97% OTUs) or UNOISE (denoising)?
The UNOISE algorithm performs error-correction ( denoising ) on amplicon reads. It is implemented in the unoise3 command .
The original UNOISE algorithm was briefly described in Edgar & Flyvbjerg (2015) . An improved algorithm was described and validated in Edgar 2016 . The implementation in unoise3 and uchime3_denovo is quite similar to UNOISE2 except for a change in parameters for chimera detection, which I believe greatly reduces the number of false positives over the original parameters described in the UNOISE2 paper that were implemented in the earlier unoise2 and uchime2_denovo commands in usearch v9.
The algorithm is designed for Illumina reads, it does not work as well on 454, Ion Torrent or PacBio reads .
Correct biological sequences are recovered from the reads, resolving distinct sequences down to a single difference (often) or two or more differences (almost always).
Errors are corrected as follows:
- Reads with sequencing and PCR point error are identified and removed.
- Chimeras are removed.
Abundances are calculated after denoising by generating an OTU table using the otutab command .
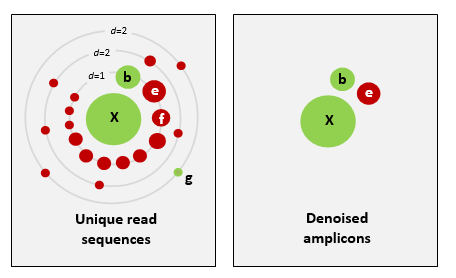
Schematic of the UNOISE2 denoising strategy (figure from the UNOISE2 paper).
The left panel shows the neighborhood close to a high-abundance unique read sequence X, grouped by the number of sequence differences (d). Dots are unique sequences, the size of a dot indicates its abundance. Green dots are correct biological sequences; red dots have one or more errors. Neighbors with small numbers of differences and small abundance compared to X are predicted to be bad reads of X. The right panel shows the denoised amplicons. Here, X and b were correctly predicted, e is an error with anomalously high abundance that was wrongly predicted to be correct, f is an error that was correctly discarded but has an abundance almost high enough to be a false positive, and g is a low-abundance correct amplicon that was wrongly discarded. The abundances of b, e, and f are similar, illustrating the fundamental challenge in denoising: how to set an abundance threshold that distinguishes correct sequences from errors.
