
OTU coverage
 See also
See also
OTU / denoising analysis pipeline
Quality control for OTUs
Comparing two sets of OTUs for the same reads
Explaining your data
Measuring coverage attempts to answer the question "what fraction of the reads is explained by my OTUs?". This gives a sense of whether the OTUs have good coverage. For example, if 95% of your reads can be explained by the OTUs, then you might feel that the coverage is good. The 5% of reads which are not explained probably contain a mix of rare biological sequences and artifacts such as sequencer errors and chimeras, but with low-abundance sequences these cannot be reliably distinguished so we have to accept some loss in sensitivity to get a reasonably low rate of spurious OTUs.
Analyzing a control sample such as a staggered mock community can be used to calibrate parameters to get the highest sensitivity with an acceptable rate of spurious OTUs.
This page describes how you can investigate coverage indepedently of control samples, e.g. because you didn't sequence any.
Classifying reads
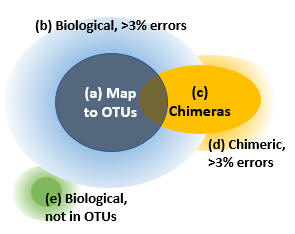
Reads can be divided into five groups as shown in the figure.
(a) Reads that map to OTUs. If we assume that the OTUs are correct biological sequences, then these reads are correct or within 3% of a correct sequence. These are the reads which are counted in the OTU table if you follow my recommendations for mapping reads to OTUs . I think it is reasonable to consider these reads explained by the OTUs, while acknowledging that there may be some cases where multiple species are lumped into a single OTU.
(b) Reads of an OTU with >3% errors. The vast majority of these will have been discarded because they do not map to an OTU.
(c) Correct reads of chimeric amplicons. Most likely, only a small minority of OTU sequences will be chimeric because the cluster_otus and unoise3 commands have very effective chimera filters. If a chimera is <3% diverged from an OTU, some or all of its reads will be mapped to that OTU. I consider this to be harmless for all practical purposes because chimeras are much less abundant than their parents, so the number of chimeric reads added to the OTU count will be small compared to other fluctuations. The large majority, or perhaps all, chimeras in your reads will have parents that are in the OTUs because chimeras are strongly biased to form between high-abundance parents, for pretty obvious reasons.
(d) Chimeric reads with errors. If a chimeric read has errors, say >3% compared with the true chimeric amplicon sequence, then it is impossible to distinguish it reliably from a noisy read of one of the parent sequences or a biological sequence missing from the OTUs.
(e) Reads of biological sequences that are not in the OTUs. We would like to estimate the size of this set, but this cannot be done reliably because the only way to do that is to estimate the sizes of (a) through (d) and see what is left over, but the uncertainties in those sizes are comparable with the size of (e). The calculation ends up being something like (e) = 100% minus (90% +/- 10%), so you really don't have much of an idea how big (e) is -- it could be anywhere between 0% and 20%.
Accounting for reads which do not map to an OTU at 97% identity
Make a FASTQ file for the unmapped reads by using the -notmatchedfq option of the otutab command .
We can safely assume that a large majority of low-quality reads which did not map belong to the OTUs and chimeras formed from the OTUs. This is because the OTUs represent the most abundant template sequences, and most bad reads will be derived from those templates just as most good reads are. With this in mind, pick an expected error threshold, say 2, and consider the reads above the threshold to be explained as noisy reads of known OTUs.
usearch -fastq_filter unmapped.fq -fastq_maxee 2.0 -fastaout unmapped_hiqual.fa \
-fastaout_discarded unmapped_loqual.fa
Now let's try to find chimeric reads. Use the -chimeras option of unoise3 to get the predicted chimeric amplicons. Do this even if you're only using 97% OTUs in your analysis, because this is the best way to get an accurate set of chimera predictions.
If an unmapped read is within, say, 5% of a chimera or an OTU, then it is probably a noisy read with more errors than expected from its Phred scores. Combine the OTUs and chimeras into one database file and use this command to check.
usearch -usearch_global unmapped_hiqual.fa -db otus_chimeras.fa -strand plus -id 0.95 \
-matched unmatched_noisy.fa -notmatched unmatched_hiqual_other.fa
Now you have the number of mapped reads and counts of low quality reads and noisy reads with apparently high quality. If you add these up, it should account for most of your data.
One way (the only way?) to check for valid biological sequences that are not accounted for in the OTUs is to search the remainng reads against a large database like SILVA, e.g.
usearch -usearch_global unmatched_hiqual_other.fa -db silva.udb -strand both \
-id 0.99 -alnout unmapped_silva.aln
I specified a high identity threshold because if you use a lower threshold, say 0.97, you may get spurious alignments from noisy reads. If the identity is much below 100%, it is difficult to decide..
