
Merging (assembly) of paired reads
See alsofastq_mergepairs command
Paired read assembler benchmark results
Edgar & Flyvbjerg 2014 paper on expected errors and Bayesianassemblyassembly
FASTQ files
Quality scores
Staggered pairs
Paired read merging (assembly)
Illumina machine can generate paired reads where constructs are sequenced in both directions.
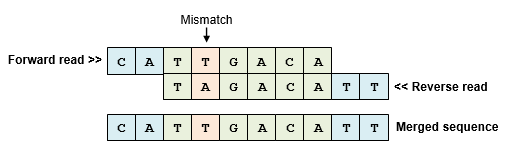
When the forward and reverse reads overlap, they can be "assembled" or "merged" to give a single sequence. I prefer to call this merging rather than assembly to distinguish from whole-genome assembly using shotgun reads. People who use the term assemblers call the result of merging a pair a "contig", I call it a "consensus sequence".
Consensus sequence
Paired read mergers generate consensus sequences by aligning the forward and reverse reads and resolving any mismatches found in the alignment. The reverse read is reverse-complemented so that its sequence is oriented on the same strand as the forward read. Usually, the alignment covers part of the forward and reverse reads leaving unaligned segments at the beginning of both reads. However, if the sequencing construct is shorter than the read length then the alignment is staggered with unaligned segments at the ends rather than the beginning of the reads. By default, the fastq_mergepairs command trims (discards) the unaligned segments if the alignment is staggered.
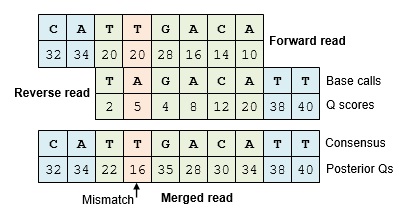
Posterior quality scores
If the base calls match, the Q score should increase. If there is a mismatch, the Q score should be usually be reduced, but not always. The Q score at a mismatch position may stay the same or even increase if one of the reads has a low enough Q score that the base call is predicted to be wrong, e.g. Q=2 which indicates an error probability of 63.1%. The error probabilities after merging are called posterior probabilities, and the corresponding Q scores are called posterior Qs. The term "posterior" comes from Bayesian statistics, meaning after new evidence has been taken into account. The equations for calculating posterior Qs are given in Edgar & Flyvbjerg (2014) .
Calculating correct posterior Qs is important because it enables an improved estimate of the number of errors in the read.
Other programs calculate incorrect posterior Q scores
Most assemblers generate posterior Q scores which are obviously wrong in at least some situations (see benchmark results ). The best (i.e., worst) example is PANDAseq, which reports lower Q scores in most aligned positions even if there is agreement between the two reads (tested through v2.8).
