
FASTQ files
See alsoQuality scores
Average Q is a bad idea!
FASTQ format options
Wikipedia article on FASTQ
Expected errors
Cock et ail (2010) paper describing FASTQ
FASTQ files are text files containing sequence data with a quality (Phred) score for each base, represented as an ASCII character. The quality score is an integer (Q) which is typically in the range 2 - 40, but higher and lower values are sometimes used. In particular, versions 1.8 and later of the Illumina platform generate reads with Q scores up to 41.
Unfortunately, the FASTQ format is not standardized. There are several variants in common use, and it is not possible to distinguish them automatically with high reliability. The fastq_chars command can be used to guess the format of an unknown file. See FASTQ format options .
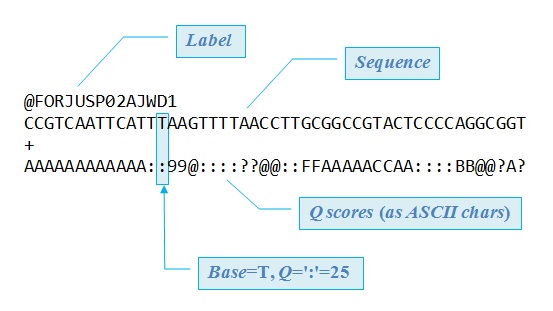
FASTQ read with 50 base calls in Illumina format (ASCII_BASE=33).
There are always four lines per read. The first line starts with '@', followed by the label.
The third line starts with '+'. In some variants, the '+' line contains a second copy of the label.
The fourth line contains the Q scores represented as ASCII characters.
