fastq_eestats2 command
See also
FASTQ files
Expected errors
The fastq_eestats2 command creates a summary report showing how many reads will pass an expected error filter at different length thresholds. This is useful for choosing parameters for fastq_filter or fastq_mergepairs, especially for reads which vary in length such as 454.
The report is written to stderr and optionally to a text file specifed by the -output option.
Lengths to show are set by the -length_cutoffs option which specifies three integers separated by commas giving the shortest length, longest length and length increment. An asterisk (*) indicates no upper length limit. Default is 50,*,50 which means that length cutoffs of 50, 100, 150 ... maximum read length will be used.
Expected error cutoffs to use are set by the -ee_cutoffs option which is given as one or more floating-point values separated by commas giving a list of the cutoffs to use. Default is 0.5,1.0,2.0. An asterisk (*) indicates that no e.e. cutoff should be applied, so all reads of at least the given length are included.
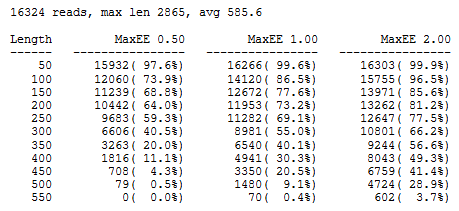
A report for a set of 454 reads is shown below. Truncating at a length of 250 looks like a a good choice because we would keep 69% of the reads with the recommended 1.0 expected error threshold, giving a good balance between read quality and keeping as many bases as possible to maximize phylogenetic resolution. The longest read is 2,865nt but the longest length shown is 550 because no reads have EE <= 2.0 when truncated at 600nt.
Note that most discarded reads will probably successfully map to an OTU
sequence using the otutab command.
Example
usearch -fastq_eestats2 reads.fq -output eestats2.txt -length_cutoffs 200,300,10