
SEARCH_16S algorithm
See also
SEARCH_16S paper
search_16s command
The SEARCH_16S algorithm searches for 16S genes in long sequences such as chromosoms and contigs. It identifies segments with a high frequency of 13-mers in known 16S genes (signature words ) , then searches within each such segment for conserved motifs close to the beginning and end of the gene. Finding a pair of motifs within the expected length range confirms the presence of the gene and provides consistent, homologous endpoints. It would be preferable to identify the true endpoints of the functional sequence, but the 16S gene is spliced out of the ribosomal operon by mechanisms that are not fully understood and lacks known sequence signals analogous to start and stop codons for protein-coding genes. I validated SEARCH_16S on finished prokaryotic genomes and curated SSU databases, finding that it has >99% sensitivity to known genes and no unambiguous false positives in control datasets containing metazoan sequences and random sequences. Details are in the paper .
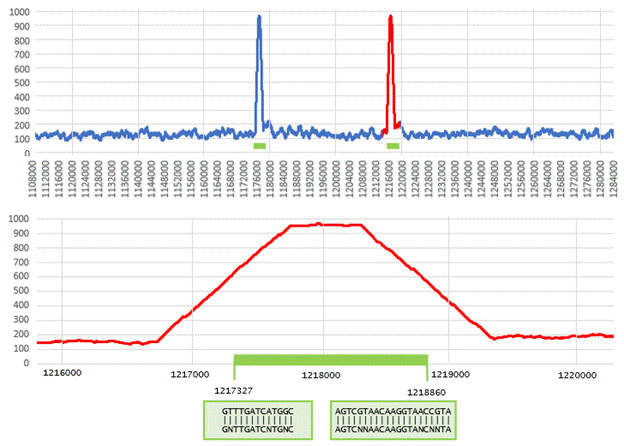
SEARCH_16S identifies two genes in a region of the E. coli chromosome reverse strand . (Figure from SEARCH_16S paper ).
In the top panel, the density of signature 13-mers over windows of length 1,000bp is shown for positions 1,108,000 - 1,284,000 in Genbank sequence AP009048.1. Most positions have a density close to the expected background of ~120 words per window. The two 16S genes in this region (green bars) are visible as spikes where the density approaches 1,000. The lower panel shows the region from positions 1,216,000 to 1,220,000 where the second gene is located. The trapezoidal shape of the density is explained by windows which contain some words before / after the beginning / end of the gene; the flat peak of length approx. 500bp is due to windows that contain only 16S words. The boundary motifs are found at positions 1,217,327 (C11F) and 1,218,860 (C1512R).
