
 Recommended taxonomy databases
Recommended taxonomy databases
See also
Microbial taxonomy
Sequence databases with taxonomy classifications
Taxonomy annotation errors in large databases
Taxonomy database downloads
Use a small database with authoritative classifications
I recommend using a authoritatively classified sequences, e.g. for 16S the most recent RDP training set or LTP release.
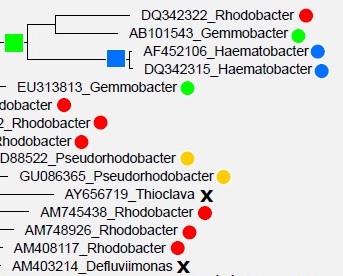 Taxonomy annotations in large databases are unreliable predictions
Taxonomy annotations in large databases are unreliable predictions
The taxonomy annotations in the large 16S databases (SILVA, Greengenes, or the full RDP database) are mostly computational predictions from 16S sequences. Roughly one in five of these predictions are wrong , probably because the guide trees have pervasive branching order errors . Therefore, using annotations from large databases adds a substantial error rate in the reference dataset on top of the intrinsic error rate of a prediction algorithm such as SINTAX or the Naive Bayesian Classifier . With these considerations in mind, I believe it is best to use a database of type strain and isolate sequences rather than Greengenes, SILVA or RDP.
References (please cite)
R.C. Edgar (2016), SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences, https://doi.org/10.1101/074161
• SINTAX taxonomy prediction algorithm
• Fast and simple method, accuracy comparable to RDP Classifier
R.C. Edgar (2018), Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences, PeerJ 6:e4652
• Cross-validation by identity, novel benchmark strategy enabling realistic accuracy estimates
• Genus accuracy of best methods is 50% on V4 sequences
• Recent algorithms do not improve on RDP Classifier or SINTAX
R.C. Edgar (2018), Taxonomy annotation and guide tree errors in 16S rRNA databases, PeerJ 6:e5030
• Approx. one in five SILVA and Greengenes taxonomy annotations are wrong
• SILVA and Greengenes trees have pervasive conflicts with type strain taxonomies
