
Abundance and amplification bias in amplicon sequencing
See also
UNBIAS algorithm
unbias command
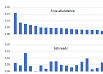 Read abundance does not correlate with species abundance
Read abundance does not correlate with species abundance
Using mock community sample with species abundances determined independently by shotgun sequencing, I found that 16S amplicon read frequencies have no meaningful correlation with species frequencies (Pearson coefficient r close to zero). Click here for figure . The factors described below cause read frequencies to diverge substantially from species frequencies.
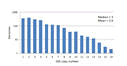 Gene copy number
Gene copy number
Prokaryotic genomes contain varying numbers of 16S genes ranging from one to ten or more, and strains with more genes therefore tend to be more common in the reads. Click here for figure .
Primer mismatches
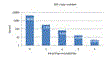 PCR amplification efficiency is strongly degraded if a template has mismatches with the primers, causing the number of reads to be suppressed, typically by an order of magnitude or more for each mismatched position. With the currently popular V4 primers, ~9% of species have one or more mismatches. Click here for figure .
PCR amplification efficiency is strongly degraded if a template has mismatches with the primers, causing the number of reads to be suppressed, typically by an order of magnitude or more for each mismatched position. With the currently popular V4 primers, ~9% of species have one or more mismatches. Click here for figure .
Sequence composition
GC content and homopolymers affect polymerase efficiency.
Sequence length
Shorter sequences amplify more efficiently. Currently popular 16S tags such as V4 have well-conserved lengths, but other markers such as fungal ITS are more variable and therefore have stronger amplification biases.
Degenerate primers
When degenerate primers are used, as is commonly the case in 16S sequencing, biases occur due to unevenness in the oligonucleotide mixture.
Biases are amplified
Small biases in efficiency, e.g. due to uneven mixing of oligos, are exponentially amplified by the PCR reaction, leading to large biases in read counts. For example, if one sequence is amplified 10% more than another in one round, it will be 1.1 20 = 7 times more abundant after 20 rounds.
