
|
|
HMP tutorial 454 reads, Human Microbiome Project samples |
Part 1. UPARSE pipeline from reads to OTU table.
Part 2. Implementing the pipeline script
With 454 data, the first step is to make sure you understand the read structure. HMP reads start with the control sequence TCAG followed by the barcode and primer.
Making the barcodes.fa file
The pipeline needs a FASTA file containing the barcodes in which the barcode sequence labels are the sample identifiers. This is the barcodes.fa file in the misc/ subdirectory, which looks like this:
>SRS042483
TCAGCTGTACGTAG
>SRS042606
TCAGCTACGTCTGT
>SRS058387
TCAGTATCACTACG
...etc...
Here, sequence labels are SRA sample identifiers, e.g. SRS042483. I added TCAG at the start of each barcode for compatibility with the fastq_strip_barcode_relabel2.py script (next step below).
Here's how I made the barcodes.fa file. Data in HMP archives is described by LMD (library metadata) files (see HMP documentation for library metadata). LMD files are tabbed text with fields described in the info/lmd_format.txt tutorial file.
The info/SRR058098.lmd file is the LMD file for the reads used in this tutorial. This contains the information we need to make the barcodes.fa file: field 7 is the barcode sequence and field 10 is the sample identifier. The make_barcodes.bash script uses these fields to make the barcodes.fa file in a single command line using the Linux sed and tr commands. This command line uses fancy Linux tricks like regular expressions and a hack to insert newlines using tr, so don't worry if you don't understand it. The main thing here is to understand the format of the barcodes.fa file so that you can make one for your own data.
Reformatting the reads
With 454 reads, the fastq_strip_barcode_relabel2.py script is used to strip the barcode and primer and add a sample identifier to the read label. This script assumes that the read starts with the barcode and primer -- this is why I added TCAG to the barcode sequences. (This design is to support reads with different or missing control sequences).
The primer sequence is field 9 in the LMD file. We can verify that all the samples have the same primer like this:
cut -f9 info/SRR058098.lmd | uniq
CCGTCAATTCMTTTRAGT
This gives us everything we need to run the Python script; this is the command line from run_uparse.bash:
../py/fastq_strip_barcode_relabel2.py ../fq/reads.fq CCGTCAATTCMTTTRAGT ../misc/barcodes.fa Read \
| sed "-es/barcodelabel=/sample=/" > $out/relabel.fq
The sed command changes barcodelabel= to sample=. This is because I now use sample=xxx as the convention for the sample identifier. Some older scripts such as fastq_strip_barcode_relabel2.py use barcodelabel= for not very good reasons.
Quality filtering and length trimming
454 reads often have highly variable lengths with quality that falls towards the ends of the reads. To get good OTUs, the reads must be trimmed to a fixed length and filtered for quality. The fastq_eestats2 provides a readable report to assist you in choosing which length to use. The command line in run_uparse.bash looks like this:
$usearch -fastq_eestats2 ../fq/reads.fq -output $out/eestats2.txt
The output file is out/eestats2.txt:
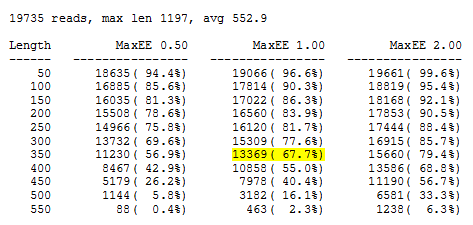
The recommended expected error cutoff is 1.0 (middle column). I chose 350 as the length (yellow highlight) because this keeps two thirds of the reads while increasing the length to 400 discards almost half. This gives the fastq_filter options -fastq_maxee 1.0 and -fastq_trunclen 350 used in run_uparse.bash (below). Don't worry if discarding one third of the reads sounds like a lot; most of the discarded reads will map to OTUs.
$usearch -fastq_filter $out/relabel.fq -fastq_maxee 1.0 -fastq_trunclen 350 \
-fastaout $out/filtered.fa -relabel Filt -log $out/filter.log