See also
UPARSE pipeline
Read quality filtering
FASTQ format options
Quality scores
Global trimming
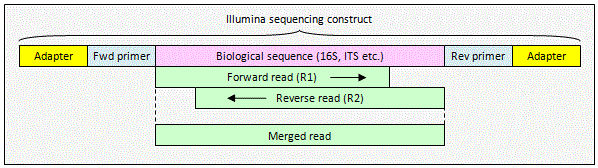
Illumina paired read with overlap
The reads contain biological
sequence only. The pair can be merged by
fastq_mergepairs to obtain a consensus biological sequence.
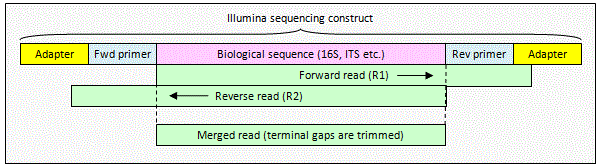
Illumina paired reads with staggered overlap
When the alignment
is staggered, one or both reads extend into non-biological sequence. The pair can be merged by
fastq_mergepairs to obtain a consensus biological sequence. The
non-biological sequence will be deleted automatically because
fastq_mergepairs detects staggered alignments and deletes terminal gaps
before building a consensus sequence.
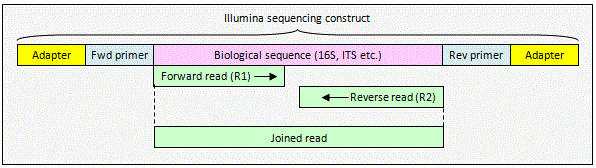
Illumina paired reads with no overlap
The reads contain
biological sequence only. The reads can be combined using
fastq_join which inserts a spacer (default
8 Ns) between the reads. OTUs can be constructed from joined reads by
dereplicating (derep_fulllength command)
and clustering with cluster_otus. This
is valid even if some of the reads overlap, giving you an option for
processing paired reads where varying amplicon length means that you
sometimes get an overlap but not always, as it typically does with ITS. For
analysis that can't deal with joined sequences you can trim to the end of
the first read using fastx_truncate.
Taxonomy prediction with utax works fine with
joined sequences because any duplicated sequence in an overlap segment will
only count once to the unique words in the sequence.
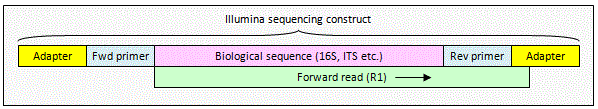
Illumina unpaired read
An unpaired read may extend into
non-biological sequence at the end of the sequencing construct. This will
happen if the read length is longer than some of your amplicons. To remove
non-biological sequence, you can use the
search_oligodb command to find the reverse primer. You will have to
write your own script to trim to the primer local as usearch currently does
not have a command for this.

454 read
A typical 454 read
starts with a control sequence (usually TCAG), followed by the barcode and
forward primer. Sometimes the read extends through the reverse primer. I
provide a python script
fastq_strip_barcode_relabel.py to extract the biological sequence from
this type of read. This script does not find or remove the reverse primer,
but that usually doesn't matter because the reads will be trimmed to a fixed
length before clustering which should delete any reverse primer sequences
with 16S at least (with ITS it may be more complicated due to the greater
variation in amplicon length).