| See also Read quality filtering fastq_filter Choosing FASTQ filter parameters Global trimming for ITS Global trimming addresses a problem that occurs in next-generation amplicon sequencing. The issue is related to terminal gaps in cluster alignments. I'll use 16S as an example since this is the most common application where it arises, but similar considerations apply for most amplicon sequencing applications. Recommendations are summarized in the table, with explanation below.
Reads should be globally alignable with NO
terminal gaps
Assessing read quality
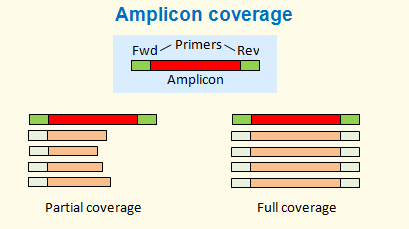
Typical 16S reads are derived from amplicon sequences. Amplicons are obtained by PCR from a pair of primers. It is important to consider whether the reads cover full or partial amplicons. Full coverage is typically obtained from overlapping paired reads.
In both cases, read lengths vary. With full coverage reads, lengths vary primarily because amplicon lengths vary (due to hypervariable regions in the gene). Minor variations can also occur due to indel errors in the reads (common in pyrosequencing reads due to homopolymers, but very rare with Illumina). With partial coverage reads, lengths vary primarily due to quality trimming. Since quality tends to fall towards the end of the reads, the last bases tend to be less reliable. This can produce an alignment with unreliable bases towards the end, as shown in the figure below.
|