See also
UPARSE-REF algorithm
UPARSE home page
OTU benchmark results
UPARSE pipeline
cluster_otus command
Introduction
The UPARSE-OTU algorithm constructs a set of OTU representative sequences from
NGS amplicon reads. It is implemented in the
cluster_otus command. Reads should be pre-processed to overlap paired reads (if
appropriate), strip barcodes, perform quality filtering and
global trimming. Post-processing is
needed to map reads to OTUs and construct a OTU table. See
UPARSE pipeline for detailed discussion of
practical issues. This page describes the OTU clustering algorithm itself.
Input sequences
Input to UPARSE-OTU is a set of sequences. Each sequence is marked with an
integer value indicating its abundance. In practice, the abundance is usually
the number of reads having a given unique sequence, but it could also be the
predicted abundance of an amplicon after a denoising step.
Clustering criteria
The goal of UPARSE-OTU is to identify a set of OTU representative sequences
(a subset of the input sequences) satisfying the following criteria.
1. All pairs of OTU sequences should have < 97% pair-wise sequence identity.
2. Chimeric sequences should be discarded.
3. All non-chimeric input sequences should match at
least one OTU with ≥ 97% identity.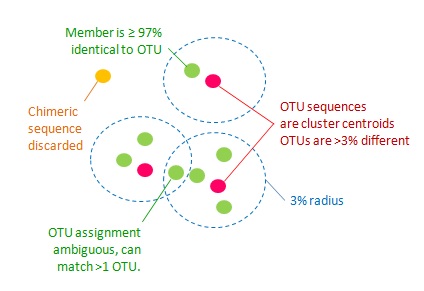
Greedy clustering
In practice, there will be a huge number of possible sets of OTUs that
satisfy the clustering criteria. UPARSE-OTU uses a greedy algorithm to find a
biologically relevant solution, as follows. Since high-abundance reads are more
likely to be correct amplicon sequences, and hence are more likely to be true
biological sequences, UPARSE-OTU considers input sequences in order of
decreasing abundance. This means that OTU centroids tend to be selected from the
more abundant reads, and hence are more likely to be correct biological
sequences.
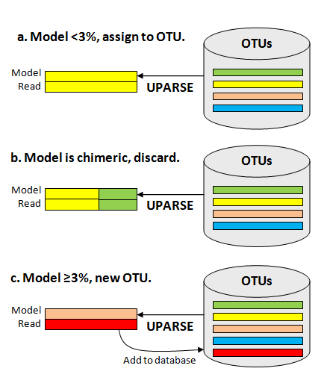
Each input sequence is compared to the current OTU database, and an maximum parsimony model of the sequence is found using UPARSE-REF (figure below). There are three cases. (a) The UPARSE-REF model is ≥ 97% identical to an existing OTU, (b) the model is chimeric, or (c) the model is <97% identical to any existing OTU. In case (a), the input sequence becomes a member of the OTU. In case (b), the input sequence is discarded. In case (c), the input sequence is added to the database and becomes the representative sequence (centroid) of a new OTU.
Reference
Edgar, R.C. (2013) UPARSE: Highly accurate OTU sequences from microbial amplicon reads,
Nature Methods [Pubmed:23955772,
dx.doi.org/10.1038/nmeth.2604].