See also
Taxonomy benchmark home
Defining "accuracy" of a taxonomy classifier
Taxonomy classification errors
Taxonomy prediction confidence measure
UTAX algorithm
RDPC often reports higher confidence than UTAX
The RDP Naive Bayesian Classifier (RDPC) has a strong tendency to "overclassify",
i.e. to give high confidence to a prediction when in fact the sequence belongs
to a novel taxon, as shown in the overclassification error
benchmark results. This is easy to test in a benchmark by leaving known taxa
out, but hard to prove on real data.
As a complement to a benchmark test, I think it is helpful to manually analyze individual cases on real data. Here, I work through a typical example as a case study.
Is this example cherry-picked?
Algorithms tend to do
well on benchmarks created by their authors. Algorithms also tend to do well
on examples picked by their author. So you would be right to ask whether
this example is typical or special. I chose it because I thought the
analysis was especially clear. So in that sense, yes, it was cherry-picked.
But it is not unusual -- I have looked carefully at many examples like this
to convince myself that the RDP results are not an artifact of my benchmark
design and it really does have a serious problem with overclassification.
A high-abundance read from mouse
feces
The query sequence (OTU_4 below) is 252nt. It was obtained from
sample data
analyzed in the mothur MiSeq SOP, which are MiSeq PE reads of the V4 region in mouse feces. There are
6,683 reads which have exactly this sequence. It is the fourth-most abundant
sequence in the reads, which means that it is almost certainly a correct
biological sequence -- we can rule out read errors and PCR chimeras, which are the
most common causes of incorrect sequences, and there is no mechanism I know of
which could cause a bad sequence to be this close to the top of the abundance
curve.
>OTU_4
CCTGTTCGATCCCCGCACTTTCGTGCCTCAGCGTCAGTAGGGCGCCGGAAGGCTGCCTTCGCAATCGGGGTTCTGCGTGA
TATCTATGCATTTCACCGCTACACCACGCATTCCGCCTTCTTCTCGCCCACTCAAGGCCCCCAGTTTCAACGGCCGGACG
GGGTTGAGCCCCGAATTTTTACCGCTGACTTAAAAGCCCGCCTACGCACCCTTTAAACCCAATAAATCCGGATAACGCTC
GCATCCTCCGTA
RDP and UTAX predictions for OTU_4
Predicted taxonomies are shown in the table below. RDPC and UTAX agree on
the prediction, but strongly disagree on the confidence. RDP training
set #14 (trainset14_032015) was used for both algorithms.
|
Rank |
Name |
RDP bootstrap |
UTAX confidence |
|
Genus |
Barnesiella |
84% |
4% |
|
Family |
"Porphyromonadaceae" |
96% |
33% |
|
Order |
"Bacteroidales" |
98% |
50% |
|
Class |
"Bacteroidia" |
98% |
73% |
|
Phylum |
"Bacteroidetes" |
100% |
99% |
The disagreement is dramatic -- RDPC reports 84% confidence (bootstrap) in the genus prediction, which is above the 80% threshold recommended by RDP, while UTAX has only 4% confidence that the prediction is correct. RDPC is even more confident at family level, reporting 96% vs. only 33% for UTAX. Manual analysis shows that the UTAX confidence estimates are more reasonable.
Guidelines for manual analysis
Manual analysis starts by
looking at alignments to a reference database with known taxonomies. The
taxonomy of the query will be the same as the top hit(s) if:
1. The identity of the top hit is HIGH, and
2. The top hits AGREE on the taxonomy.
How high is high enough for the identity? With full-length 16S sequences, we need to see an identity of around 97% or above to believe the species is the same. With short NGS reads, the species is often different even when the identity is >97%. If the identity is below around 95%, then the genus is often different. If we see a two or more different names for a given rank in the top hits, then we know that the rank cannot be reliably predicted from the hits.
Majority rule?
Some people have argued that if we see a
majority name at a given rank (say, more than half or more than two thirds),
then this is the best prediction. For example, this strategy is used by the
QIIME assign_taxonomy.py -m uclust method (the default) and by GAST. These
rules do not work well in practice because the majoriries are usually due to
strong sampling biases in the database rather than inherent properties of
different clades.
Top hits for the example sequence
The table below shows the top six hits for our example sequences according to
usearch_global.
|
Hit |
%id |
RDP Accession |
Family |
Genus |
|
#1 |
87.0% |
AB238922|S000650586 |
"Porphyromonadaceae" |
Parabacteroides |
|
#2 |
85.8% |
AB370251|S001093530 |
"Porphyromonadaceae" |
Barnesiella |
|
#3 |
85.4% |
AB267809|S000734936 |
"Porphyromonadaceae" |
Barnesiella |
| #4 | 84.2% | AB260026|S000804233 | Bacteroidaceae | Bacteroides |
|
#5 |
83.8% |
AB238928|S000650592 |
"Porphyromonadaceae" |
Parabacteroides |
|
#6 |
83.4% |
X82823|S000013923 |
"Porphyromonadaceae" |
Porphyromonas |
Notice that the top hit is below 90% and belongs to a different genus, Parabacteroides. The relatively low identity and different genus in the top hit means that our confidence that the true genus is Barnesiella should be low. Note that different programs will often report different identities for the same pair of sequences. This is especially true when identity is low. Also, identity is not a very good measure of evolutionary distance because all differences are equally weighted, while deletions are typically less likely than substitutions and the rate varies by region (e.g. hypervariable vs. constant). So the identities and order of the hits should not be taken too seriously. However, finding four genera and two families with similar identities among the top six hits clearly shows that an identity of around 85% to OTU_4 is not high enough to determine the genus or family, at least on the basis of sequence identity.
Nearest neighbors
The nearest-neighbor table from the
utaxalnout file for OTU_4 is shown below (full
report here).
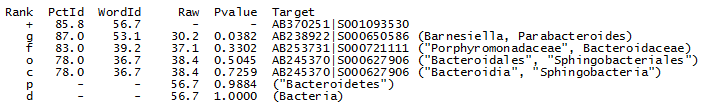
UTAX ranks hits by word identity (WordId, the number of words in common between the query and target sequence) rather than alignment identity (PctId) because WordId is much faster to calculate and gives results that are just as good on my benchmark tests, reflecting that both measures correlate more or less equally well with taxonomic rank. From the table we can see that the top hit by word identity (AB370251|S001093530) has lower alignment identity than the nearest neighbor at genus level. In other words, the closest genus by word identity is different from the closest genus by alignment identity. This again shows that other genera have indistinguishable identities to the top hit, confirming that the genus prediction should have low confidence unless we have convincing evidence from a different type of analysis.
The nearest neighbor at family level has only a small difference (83% vs. 86% or 87% for the top hit). This is only slightly better than genus -- the difference could be due to different evolutionary rates in different families, missing data or imperfect correlation between identity and taxonomy. This analysis supports the low family confidence reported by UTAX.
Bayesian magic?
So far, we have seen that sequence identity is clearly not enough to predict
genus or family with confidence. Is it possible that the Naive Bayesian
Classifier algorithm can detect a signal we can't see from the alignment
identities?
A first place to look is the training examples. The training set has just two sequences for Barnesiella: AB267809|S000734936 and AB370251|S001093530. These are 95% similar over the V4 region, so they are more similar to each other than they are to the OTU_4 sequence (85% and 87%). Given only two sequences that are 95% identical, it is hard to imagine how an algorithm could figure out which of the 8-mers in those sequences are predictive. This situation is not unusual -- the RDP training set has 2,799 different taxa of which 1,133 (40%) have only one reference sequence Actually, most taxa have no training sequences! Conceptually, the classifier needs to identify 8-mers that have higher probability in a given genus (say, Barnesiella) compared to other genera, but no algorithm can do this reliably if most genera are missing from the database.
In fact, the Bayesian statistical method used by RDPC gives obviously wrong answers. The theory is used to calculate the probability (technically called a posterior) that a sequence belongs to a given taxon. The posteriors are obviously wrong because they are always tiny -- millionths or billionths instead of being close to 1.0 for correct predictions. If the theory worked reasonably well, a Bayesian classifier algorithm would report the posteriors for each predicted taxon. Bootstrapping is used instead because the probabilities are wrong. Bootstrapping reports how often some other taxon has the highest posterior at a given rank when words are randomly subsampled. This is effectively a measure of the distance between the most probable and second most probable taxon, not of the probability that the top taxon is correct. If the training set was complete or almost complete, this would be a reasonable procedure, but with a sparse reference database you may get a high bootstrap value because the reference sequence doesn't have any known close relatives, which could be due to missing data.
It's difficult to say exactly why RDPC gives such high confidence at genus
and family level because the algorithm is opaque -- for all practical
purposes, it is impossible for a user to understand and check the evidence
used to make a prediction. By contrast, UTAX makes its prediction on the
basis of sequence similarity (measured by word identity) and the distance to the nearest neighbor at each
rank. This evidence is shown in the nearest neighbor report and can be
reviewed manually. In my opinion, the ability to understand and review the
evidence supporting a prediction is an important advantage of UTAX.