
Cross-validation by identity
See also
TAXII benchmark
Microbial taxonomy
sintax command
nbc_tax command
Top-hit identity distribution
distmx_split_identity command
Cross-validation by identity (CVI) is a benchmark strategy for assessing accuracy of predictions of features or traits from genes or gene fragments such as 16S sequences. In the TAXXI benchmark , it was used to measure accuracy of taxonomy prediction.
Motivation
CVI explicitly models varying distances between query sequences and the reference database. This is because in practice, OTUs have a range of identities with the reference. OTU identities are sometimes low, creating a challenging problem for prediction algorithms. For example, with taxonomy prediction, only ~20k 16S sequences are currently known from isolate strains with reliably known taxonomies, and OTUs often have low identities (say, 90% and lower) with these sequences. Prediction is more difficult at lower identities, but in most previous benchmark tests query sequences usually have high identities with the reference and thus fail to model a realistic scenario. The range of identities between a given set of OTUs and a reference database can be summarized as a top-hit identity histogram .
 Implementation
Implementation
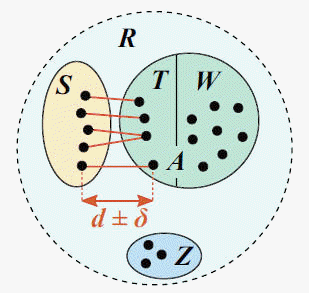
The distmx_split_identity command implements CVI is as follows. A reference database with known traits (e.g., taxonomy) is split into test and training sets such that for all test sequences, the most similar training sequence has a given identity (top hit identity, d ), e.g. d =97% (see figure). R is the reference database, which is divided into disjoint subsets S , T , W and Z . S is the test set; the training set is A = T + W . T is the set of top hits for sequences in S , which are constrained to be in the range d +/- delta where delta specifies the maximum
allowed deviation from the desired identity d . W contains reference sequences with identity < d ; these are retained to create the largest possible training set. Z contains sequences which cannot be assigned to S , T or W without violating the identity constraint.
Making a benchmark dataset
Construct test-training pairs for several different identities. This enables assessment of accuracy at varying distances from the reference. For example, with taxonomy, high accuracy at family rank is expected for query sequences having 100% identity with the reference database, but lower accuracy at 90%; these expectations can be validated by test/training pairs at 100% and 90% identities, respectively. Query sequences belonging to novel taxa, i.e. taxa not found in the reference, are modeled in test/training pairs with d < 100%. For example, most pairs of 16S rRNA sequences in a given genus have >= 95% identity. Therefore, with d = 90% most test sequences will belong to genera which are absent from the training set, and with d = 95% there will be a mix of present and absent genera. Thus, novel taxa arise naturally by construction of the test/training pairs, and the frequency of novel and known
taxa at each rank is determined by identity, which can be measured for any OTU, rather than taxonomy, which is not known.
References (please cite)
R.C. Edgar (2018), Accuracy of taxonomy prediction for 16S rRNA and fungal ITS sequences, PeerJ 6:e4652
• Cross-validation by identity, novel benchmark strategy enabling realistic accuracy estimates
• Genus accuracy of best methods is 50% on V4 sequences
• Recent algorithms do not improve on RDP Classifier or SINTAX
R.C. Edgar (2018), Taxonomy annotation and guide tree errors in 16S rRNA databases, PeerJ 6:e5030
• Approx. one in five SILVA and Greengenes taxonomy annotations are wrong
• SILVA and Greengenes trees have pervasive conflicts with type strain taxonomies
