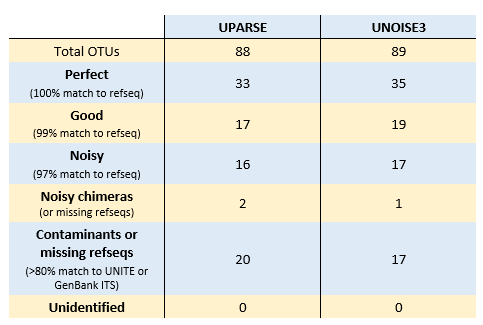
UPARSE and UNOISE3 analysis of Brown 2017 mock data
Download detailed results and scripts: brown_2017_analysis.tar.gz (requires usearch v10.0). Reads can be downloaded from SRA SRR3722592 .
High-quality OTUs were obtained with full-length sequences obtained by paired read merging. I deleted primer-binding sequences because PCR tends to cause substitution errors in those positions. I did not truncate to a fixed length or remove 5.8S or LSU sequences. These results therefore contradict Brown's conclusion that sequences should be truncated. Also, I did not use ITSx to identify ITS2 sequences; in my opinion this is an unnecessary step which could introduce false negative errors.
The results are difficult to interpret because many of the reference sequences are not found in the reads. I suspect that there are biological variations in the mock species that were not captured by Sanger sequencing. If so, there is no conclusive evidence that any of the sequences reported by UPARSE or UNOISE3 are incorrect and in fact all of the sequences could be perfect.
According to Brown2017, there are 180 species in the mock community. There are 163 reference sequences (GenBank KU535697-535859) for 162 species (Ascomycota sp. L310 has two refseqs). 104 of the refseqs match the reads at 97% identity, 44 have at least one match at 100% identity. Two refseqs match exactly one read at 100% identity; these will therefore be singleton uniques and will be discarded. The read quality is very good (mean expected errors 0.1 after merging), so I think these numbers are most likely explained by two issues: many species are missing from the reads, and some of the reference sequences have differences relative to the strains sequenced in the MiSeq run. One possibility might be intra-species variation which was not captured by the Sanger sequencing.
I ran UPARSE and UNOISE3 following recommended procedures . I used uparse_ref to classify OTUs and ZOTUs by comparison with the refseqs; results shown in table below. I think most of the "Good" and "Noisy" sequences are probably explained by incorrect or missing refseqs and the OTU sequences are in fact correct. Since only 44 of the refseqs have exact matches in the reads, an OTU clustering method can identify at most 44 "perfect" sequences when assessed by comparison with the refseqs. Both of the putative chimeras had differences relative to the model, and I think these are quite likely to be correct biological sequences which have "fake models" (see UCHIME2 paper ).
OTU73 and Zotu85 are identical and do not match UNITE at 80% identity. However, they match GenBank KT243473.1 (uncultured fungus ITS)with 93% identity. All other OTUs and ZOTUs either match a reference sequenc with >97% identity or a UNITE sequence with >80% identity.